Синдром ломкой Х-хромосомы (FXS, синдром Мартина-Белл)
Это наследственное Х-сцепленное (связанное с полом) заболевание, возникающее в результате нарушений в гене FMR1 (fragile X mental retardation 1). Синдром характеризуется умственной отсталостью разной степени, нарушением концентрации внимания, речевого развития, а также может сопровождаться другими клиническими проявлениями.
Синдром ломкой X-хромосомы может встречаться у обоих полов, но мужчины страдают этим заболеванием чаще и тяжелее, так как имеют всего одну X-хромосому.
Распространенность: 1 на 4000 среди мужчин и 1 на 6000 среди женщин.
Что означает «ломкость»
X-хромосомы?
«Ломкость» Х-хромосомы заключается в её особом строении.
В ходе цитогенетических исследований было обнаружено, что при введении фолиевой кислоты в клеточную культуру некоторых пациентов с умственной отсталостью в небольшом проценте клеток у длинного плеча одной из X-хромосом, в котором располагается мутировавший ген FMR1, краешек как бы отшнуровывается, повисает на ниточке.
Однако данный метод недостаточно чувствителен и на данный момент редко используется для диагностики синдрома.
Генетическая основа данного явления — это увеличение числа тринуклеотидных повторов (то есть увеличение количества нуклеотидов CGG) на длинном плече Х-хромосомы.
У здоровых людей число этих повторов составляет от 6 до 45. Клиническое проявление заболевания наблюдается при числе CGG-повторов свыше 200, что считается «мутацией». Состояние, когда число CGG-повторов находится в пределах 55-200, называют «премутацией». У людей с таким количеством CGG-повторов заболевание в типичной форме не имеется, но высока вероятность того, что оно проявится у их потомков. Это обусловлено возможностью увеличения числа тринуклеотидных повторов во время оогенеза.
«Серой зоной» называют количество повторов от 45 до 54. Она не имеет никакого влияния на здоровье человека, но может приводить к проблемам у будущих поколений, как и в случае премутационного состояния гена.
Клинические признаки синдрома ломкой Х-хромосомы
Больные с данным синдромом имеют нарушения интеллектуального развития, от умеренного отставания в обучении до более серьезных проблем. Им также свойственна быстрая, сбивчивая и бормочущая речь.
У мальчиков имеются характерные фенотипические (лицевые) особенности: большая голова с высоким и широким лбом, длинное лицо с увеличенным подбородком, несколько уплощённая средняя часть лица, тупой, слегка клювовидно загнутый кончик носа и большие оттопыренные уши.
Поведенческие характеристики могут включать аутизм и аутичные черты, гиперактивность, привычка грызть руки и хлопать в ладоши, агрессивность, трудно устанавливаемый зрительный контакт. Патогномоничным признаком в постпубертатном возрасте, который заставляет заподозрить заболевание, является макроорхизм (увеличение размеров яичек) при отсутствии эндокринной патологии.
Другими характерными проявлениями являются гипермобильность суставов, частые отиты, страбизм, пролапс митрального клапана, гастроэзофарингеальный рефлюкс. Клиническая картина может сильно варьировать, может присутствовать как только один, так и несколько признаков.
Основные симптомы, наблюдаемые у мужчин, могут наблюдаться и у женщин. Однако у них преобладают не настолько выраженные интеллектуальные дефекты, умеренные поведенческие проблемы и физические признаки FXS.
Примерно треть женщин с FXS значительное отстают в умственном развитии, остальные же могут иметь умеренное отставание или некоторую неспособность к обучению, эмоциональные, психические проблемы, общее беспокойство и/или социальную тревожность.
Небольшой процент женщин с полной мутацией в гене FMR1 не имеют явных признаков интеллектуального, поведенческого или физического отклонений. FXS у них часто диагностируется только после того, как это заболевание обнаружено у других членов семьи.
Расстройства, связанные
с премутацией (55-200 повторов CGG) в гене FMR1
Первичная недостаточность яичников (FXPOI), ассоциированная с ломкой Х-хромосомой
Первичная недостаточность яичников характеризуется снижением овариального резерва, наступлением менопаузы до 40 лет и повышенным уровнем фолликулостимулирующего гормона (ФСГ).
Термин «первичная недостаточность яичников» или «преждевременная недостаточность яичников» (ПНЯ) в настоящее время применяется для определения заболевания, которое ранее характеризовалось как преждевременная менопауза, преждевременная яичниковая недостаточность или вторичный гипогонадальный гонадизм. Одним из основных отличительных признаков преждевременной яичниковой недостаточности является повышение уровня фолликулостимулирующего гормона. Большинство случаев ПНЯ являются спорадическими, частота проявления у женщин с хромосомным набором 46 ХХ составляет, в среднем, около 1%, при этом доказана тесная связь данного нарушения c возрастом.
Однако приблизительно 10-15% пациенток с ПНЯ имеют отягощенный семейный анамнез.
В этиологии ПНЯ можно выделить два основных механизма: уменьшение числа (истощение пула) фолликулов и их дисфункцию. Причины, приводящие к развитию ПНЯ, весьма гетерогенны: генетические, ферментативные, аутоиммунные, инфекционно-токсические, ятрогенные, психогенные. В последние годы большое внимание исследователей уделяется молекулярно-генетическим аспектам данной патологии яичников, поскольку выявлен определенный набор генов, который может отвечать за развитие ПНЯ. Показано, что изменения в гене FMR1, расположенном на Х-хромосоме (область Xq26-28), существенно увеличивают риск возникновения данной патологии.
Первичная недостаточность яичников, связанная с ломкой хромосомой X (FXPOI), возникает у носителей премутации (55-200 повторов CGG). У женщин со спорадическими формами ПНЯ частота премутации гена FMR1 колеблется от 0,8 до 7,5%, в то время как при семейных формах заболевания она может достигать 13%. Показано, что женщины-носительницы премутации гена FMR1 намного чаще (38%) страдают олигоменореей в сравнении с общепопуляционными данными (6%).
В данном случае наличия премутационных аллелей (~60-200 повторов) у женщины существует риск увеличения числа повторов (экспансии) у её потомков до полной мутации, который растет с величиной области повторов. Когда число повторов превышает 100, имеется систематический риск экспансии до полной мутации в следующем поколении.
Как правило, материнская премутация гена FMR1 у потомков переходит в полную мутацию, приводя к возникновению синдрома ломкой Х-хромосомы, тогда как отцовская премутация, как правило, стабильна. Во многих FMR1-аллелях содержатся последовательности AGG, разбросанные среди участков повтора CGG. Полагают, что эти AGG образуют «перерывы», которые придают ДНК устойчивость и снижают риск экспансии в следующем поколении. Профиль риска у матерей, не имеющих перерывов AGG, выше, чем у матерей с тем же числом повторов, но имеющих хотя бы один участок AGG, который уменьшает число идущих подряд последовательностей (CGG)n.
Тремор/ атаксия, ассоциированные с ломкой Х-хромосомой (FXTAS)
Также наличие премутации коррелирует с проявлением связанного с ломкой хромосомой X тремор-атаксического синдрома (FXTAS).
FXTAS – это нейродегенеративное заболевание старшего возраста, чаще свойственное мужчинам. Его первые признаки обычно возникают после 50 лет. До появления симптомов FXTAS у большинства заболевших не было каких-либо медицинских или неврологических проблем. Женщины составляют лишь небольшую часть больных FXTAS, и их симптомы, как правило, менее серьезны.
Особенности FXTAS включают:
- Мозжечковую атаксию (проблемы с равновесием).
- Интенционный тремор (динамические движения появляющиеся или усиливаюшиеся во время каких-то действий, письма и т.д.).
- Потерю памяти (как правило, краткосрочную).
- Неустойчивость настроения, раздражительность, изменение личности, психиатрические симптомы.
- Симптомы паркинсонизма (многим больным ошибочно диагностируют болезнь Паркинсона).
- Деменцию (ошибочно диагностируют болезнь Альцгеймера).
- Снижение когнитивных функций (снижение математических способностей, способности к чтению и словесных навыков, понимание).
- Прогрессирование FXTAS с разной скоростью.
Генетика и наследование
Как говорилось раннее, в 99% случаев первопричиной FXS является увеличение числа тандемных триплетных повторов CGG в 5′-нетранслируемой области гена FMR1. (Описаны случаи других мутаций в том же гене и его окрестностях, а также мутации в близком гомологе FMR2, дающие похожий фенотип, но так бывает очень редко). У всех людей ген FMR1 содержит тандемные триплетные повторы CGG в 5′-нетранслируемой области. При передаче гена потомству это число может незначительно уменьшаться-увеличиваться: в поколениях этот участок генома как бы «дышит».
Если у женщины в одной из ее X-хромосом число повторов превышает 50 (по статистике, такое имеет место в одной на 250 X хромосом, то есть очень часто), возникает риск резкого спонтанного увеличения в клетках-предшественниках ооцитов при мейозе.
Если в результате такого спонтанного увеличения число повторов превысит 200 и именно этот аллель достанется ребенку, у него этот аллель FMR1 экспрессироваться не будет вовсе.
Если этот ребенок окажется девочкой, то эффект делеции может быть в какой-то степени компенсирован работой нормального аллеля на X-хромосоме, полученной от отца, особенно при благоприятном паттерне инактивации одной из X-хромосом в соматических клетках.
Если же ребенок – мальчик, то у него FXS разовьется во всей своей тяжести.
Экспансия числа повторов проявляется в зависимости от своей степени
Носительство так называемой премутации (числа повторов от 50 до 200) может фенотипически никак не выражаться, так как ген FMR1 все-таки экспрессируется, или, как было описано выше, приводит к FXPOI у женщин репродуктивного возраста и к FXTAS у мужчин и женщин старше 50 лет.
При полной мутации (числе повторов более 200 ген не экспрессируется: на обширную экспансию повторов CGG клеточные системы реагируют метилированием промотора, т.е. вышележащего участка ДНК, где инициируется транскрипция гена. Из-за блокировки промотора ген FMR1 не экспрессируется, белок FMRP не синтезируется; он не выполняет своей функции.
В норме белок FMRP присутствует в разных органах и тканях, но особенно ярко представлен в нервной системе. Белок FMRP является важнейшим регулятором процессинга мРНК в нервных клетках. Отключение экспрессии FMR1 из-за критического увеличения числа повторов CGGв 5′-нетранслируемой области гена меняет весь профиль синтеза белков в нейронах мозга. На уровне клеточной морфологии это выражается в образовании аномально большого количества дендритных шипиков измененной формы.
Возникая в большом количестве, такие шипики не дают функционально зрелых синапсов; результат – аномальная гиперпластичность нейронных сетей, несовместимая с приемом и переработкой информации из внешнего мира в нормальном, характерном для человека режиме.
Синдром ломкой X-хромосомы может быть диагностирован по поведению ребенка к концу первого года жизни, но в первые недели и даже месяцы этого сделать, как правило, нельзя, т.е. синдром не «акушерский».
В родословных больных с синдромом ломкой X-хромосомы отчетливо прослеживается феномен антиципации. Данный феномен заключается в усугублении клинических проявлений в каждом новом поколении из-за нарастания экспансии тринуклеотидных повторов.
В связи с этим большую клиническую значимость имеют скрининг и диагностика синдрома ломкой Х-хромосомы.
Кто должен пройти диагностику на
синдром ломкой Х-хромосомы?
Генетический тест, определяющий нарушения в гене FMR1,
необходимо пройти:
- Пациентам с задержкой развития, интеллектуальной недостаточностью неизвестной этиологии или аутизмом и их родственникам.
- Женщинам с нарушениями репродуктивной функции, ассоциированными с повышенным уровнем ФСГ, олигоменореей.
- Пациентам с тремором и мозжечковой атаксией в пожилом возрасте.
- Пациентам, у родственников которых наблюдались нарушения в гене FMR1.
Как проводится тестирование на синдром
ломкой Х-хромосомы
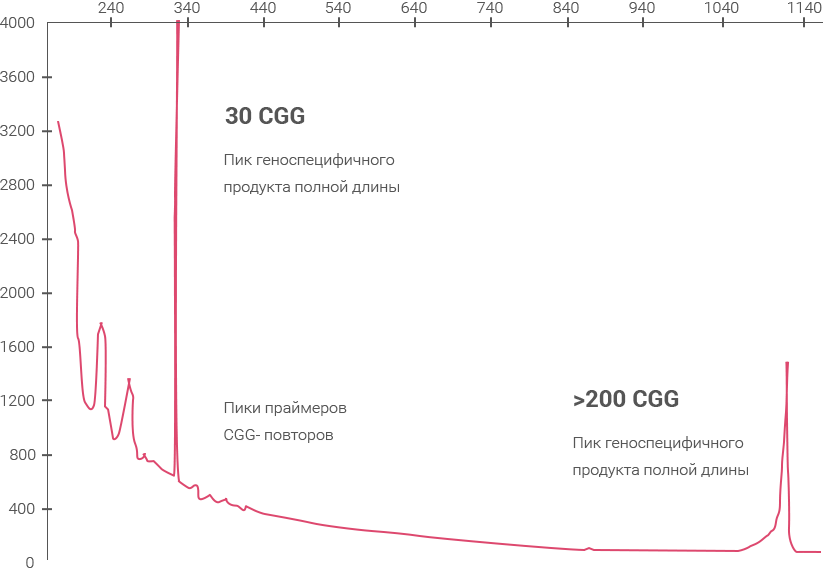
Основным методом в диагностике FXS, FX-TAS и FX-POI, а также для выявления риска экспансии повторов до полноий мутации в следующем поколении у пациентов с премутациеий используют полимеразную цепную реакцию (ПЦР) с детекцией полученных продуктов на капиллярном электрофорезе.
Анализ обеспечивает выявление премутации путем точного определения размеров аллелеий до 200 повторов CGG и идентификацию полномутационных аллелей CGG как у мужчин, так и у женщин, а также позволяет идентифицировать AGG-перерывы. Условия проведения данного теста указаны здесь
Основные принципы
диагностики
Геноспецифичная ПЦР
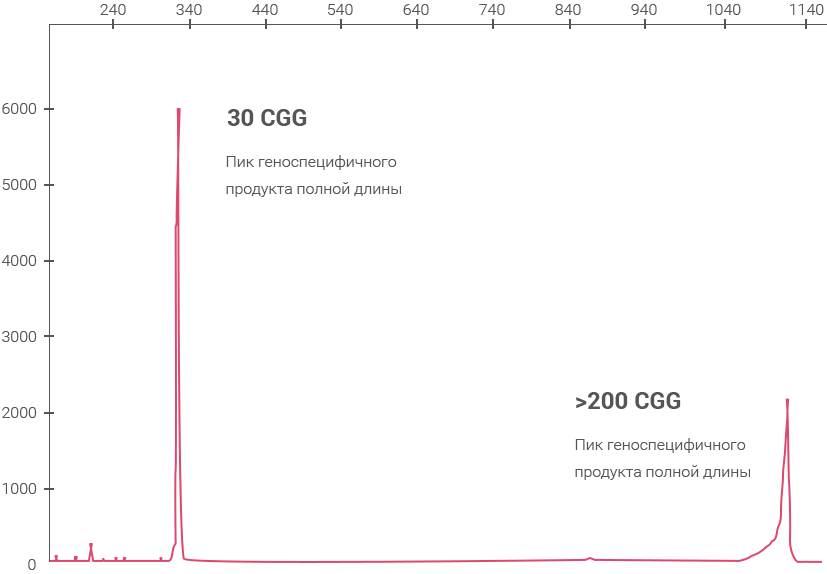
CGG RP PCR
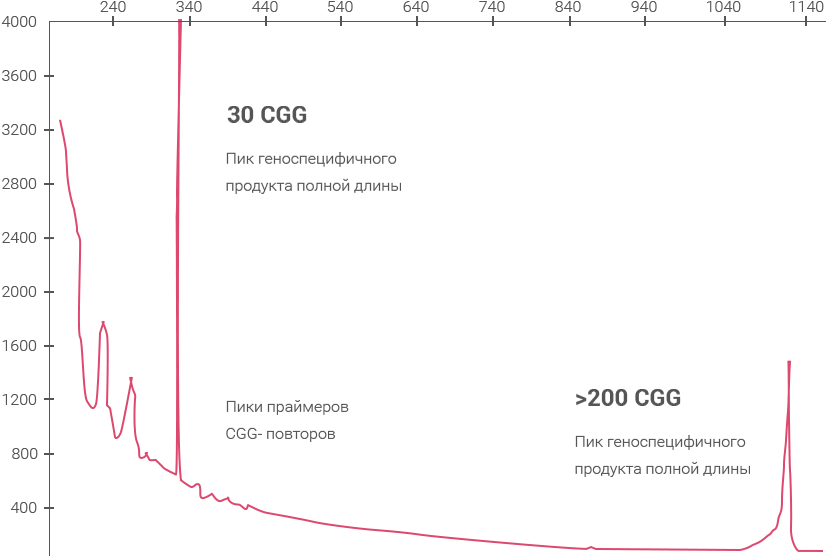
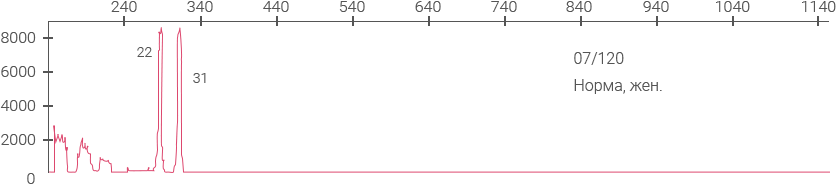
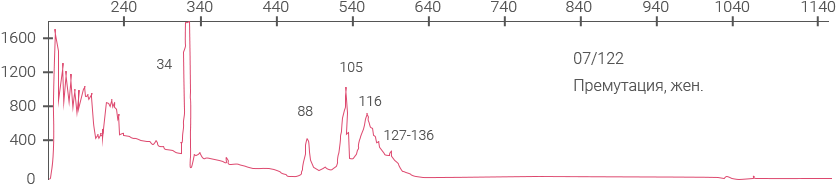
Примеры полученных
данных
Электрофореграммы панели ВОЗ для определения синдрома ломкой Х-хромосомы (NIBSC #08- 158)
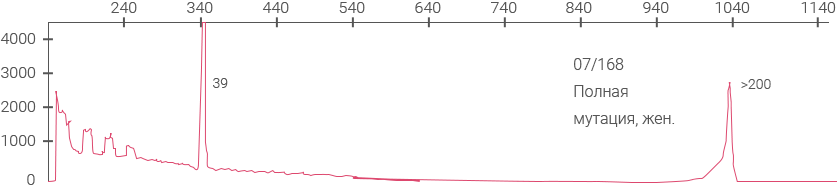
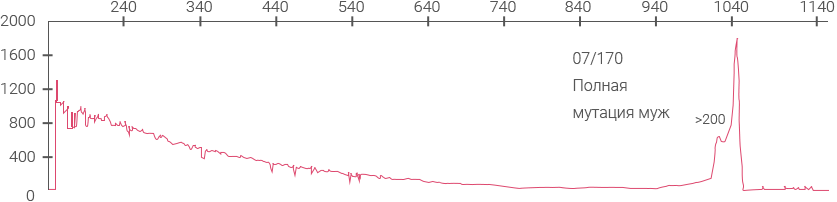
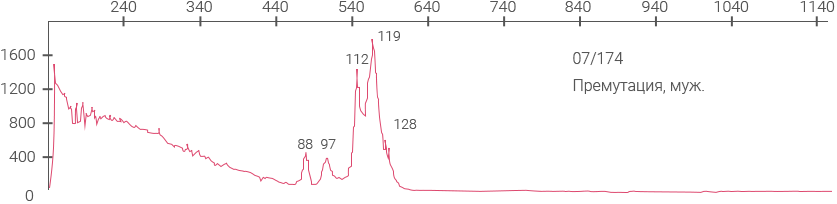
Электрофореграмма, демонстрирующая наличие AGG-перерывов на аллеях 30 и 60 CGG
Зачем диагностировать этот
синдром, если он не лечится?
С точки зрения репродуктивной медицины очень важно уметь прогнозировать рождение новых детей с FXS. Теперь это возможно!
С точки зрения коррекционной практики работу с такими детьми можно усовершенствовать: совсем недавно в США завершились клинические испытания препаратов, позволяющих смягчить ряд симптомов, в частности, улучшить социализацию (Hagermanetal, 2012).
Почему надо скринировать беременных (на как можно более ранних стадиях), а не новорожденных (в развитых странах тестирование на риск развития FXS у потомства уже включено во многие программы ведения беременности)?
Во-первых, если детектировать премутацию у потенциальной роженицы, еще можно сделать так, чтобы «больная» хромосома вообще не досталась ребенку (у женщины две X-хромосомы, она может передать ребенку «здоровую»).
Для этого существует ЭКО (либо перинатальная диагностика на ранних сроках, при неудачном раскладе можно прервать беременность).
Women tested (n= 14,334)

(<50 CGG repeats)
(n= 14,127)

(50 -199)
(n= 204)

(>200)
(n= 3)
(n= 177)

fetal allele
(n= 87)
fetal allele
(n= 85)

fetal allele
(n= 5)
H. Toledano-Alhadefetal., AmJHumGenet 69:351–360, 2001
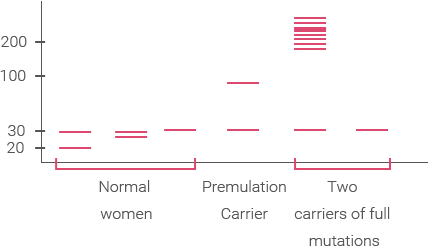
Во-вторых, с точки зрения методов молекулярной биологии с их ограничениями, детектировать премутацию проще и надежнее, нежели полную мутацию. Премутация лучше детектируется с помощью ПЦР, в то время как полная мутация >500 повторов может не дать ПЦР-продукта (у девочки с полной мутацией в одной из X-хромосом картинка может выглядеть, как будто у нее в двух хромосомах одинаковое нормальное число повторов; сравните третий и шестой образцы на схеме электрофореза):
Number of CGG repeats
Что делать после получения
результатов?
По результатам ДНК-диагностики необходима консультация врача-генетика, который сможет правильно интерпретировать полученные результаты и составить план дальнейшего ведения пациента.
В случае выявления бессимптомного носительства нарушений в гене FMR1 будут обсуждены вопросы планирования семьи и современные возможности рождения здорового ребенка.